The changing regulatory environment
Whilst nitrosamines were found in foods and beverages as early as the 1970s, with the controls that exist in the pharmaceutical world, their potential occurrence in pharmaceutical products was either not considered or not considered possible; that was until they were detected, identified, and measured. In 2018, an unknown impurity in the generic angiotensin II receptor blocker Valsartan was identified as the nitrosamine, N-nitrosodimethylamine (NDMA), a potent genotoxin in several species and a likely human carcinogen. Since then, impactful guidelines that describe the controls necessary to limit the potential generation of this class of compound have been developed and published. As more nitrosamines have been discovered, daily intake limits have been established.
A risk-based approach
All guidance documents included the demand for a risk-based approach to detecting the potential presence of nitrosamines or for nitrosation of their drug compounds. Some risks include the presence or potential for:
- Secondary, tertiary, or quaternary amines in the product, either as an active drug compound, excipient, or a synthetic/degradant impurity. An acidic environment or microenvironment in solid dose products to help potentially support the reaction if other ingredients are present and conditions are suitable for the generation of nitrosamines.
- Nitrite or nitrate that may have been present as a reagent in synthesis and present in an oxidative environment to help the formation of nitrosamines.
- Certain organic solvents, including amides, which may either contain secondary amine impurities or form them upon thermal degradation, reacting with commonly used nitrous acid to form nitrosamines. Tertiary and quaternary amine reagents are commonly used in synthesis which can potentially be converted to nitrosamines.

Guidelines were created in all major pharmaceutical jurisdictions (US-FDA, EU-EMA, UK-MHRA, and Japan-PMDA). Others, including Health Canada and Australia, chose to follow a combination of guidances (1-6), establishing some of the strictest controls and limits to protect their patient populations. Guidelines initially listed the nitrosamines and daily dose limit levels to support the risk assessments. Nitrosamine formation in pharmaceuticals was investigated, and potential causes were identified, this information was then used to guide companies to conduct risk assessments for their entire product ranges, particularly when changes were being proposed for existing and new products. The resulting investigations have revealed many more nitrosamines including Nitrosamine Drug Substance Related Impurities (NDSRI’s).
Risk Assessment is one of the important phases of the regulatory approach. This involves a review of reagents and synthetic processes for all products to identify possible nitrosamine sources from reagents or possible by-products of the synthetic process. The review should also consider any other contributing factors for nitrosamine formation. It should be understood that testing alone is not a substitute for ensuring all risks are identified and that testing is appropriate to those risks.
These risk assessments and product screening have generated unwelcome surprises for the industry. There have been many product recalls over the past four years resulting from cases where guidance specifications for the presence of known nitrosamines and NDSRI’s were not satisfied.
The table below is a summary of nitrosamines known to originate from common reagents or be produced from them (1-3, 6), in addition to other nitrosamines SGS have been asked to assess.
To read the full table download the whitepaper
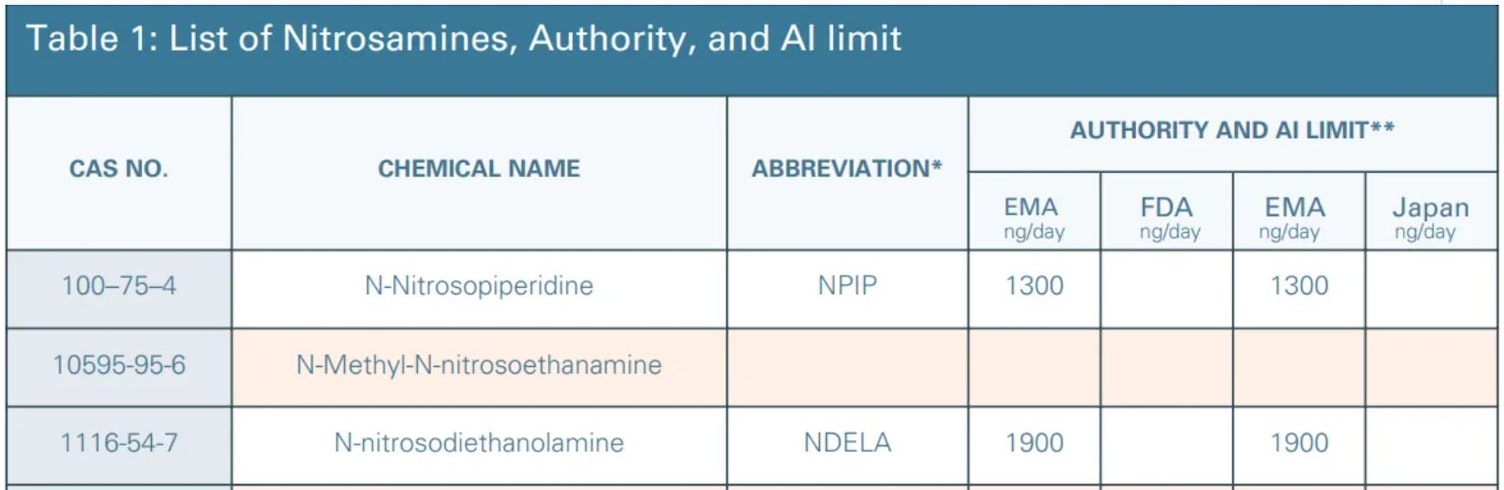
The regulators introduced a mostly harmonized specification list for nitrosamines and possible NDSRI’s. Additionally, they created a "Generic drug specification for any other Class Specific TTC" category which, until assessed or specified, has been assigned a limit of 18ng/day unless otherwise justified. It should be stressed that the limits in table 1 apply when only one nitrosamine is present. In cases where more are present, the FDA and EMA reference offers guidance as follows:
Where there was more than one nitrosamine present authorities invited companies to contact the agency.
The impact on the industry has also resulted in positive industry collaboration and the publication of "The Landscape of Potential Small and Drug Substance-Related Nitrosamines in Pharmaceuticals" in The Journal of Pharmaceutical Sciences (7). The report states; "following the in-silico analysis of more than 12,000 small molecule drugs and drug impurities[...] 40.4% of the analyzed APIs and 29.6% of the API impurities are potential nitrosamine precursors." This finding is highly significant and illustrates the magnitude of the problem with regard to the modification of potential synthetic routes.
The result of this finding required a more systemic approach to the problem. The EMA updated their Q&A document (1) and the FDA issued new guidance (11) and updated their web pages (12) to support the industry where a process is presented with; five Predicted Carcinogenic Potency Categories coupled with a chemical structural evaluation to determine a theoretical “Potency Score” based on substituents linked to the α and β carbons of the NDSRI’s relative to the N-Nitroso group. This scoring system considers structural features that increase or decrease the favorability of the α-hydroxylation mechanism of metabolic activation (13) responsible for the mutagenic and highly potent carcinogenic response observed for many -N-Nitrosamines. The flow chart with a progressive scoring system for the following helps companies evaluate the following to calculate the “feature score”
- Count of hydrogen atoms on each α carbon
- List of deactivating features
- List of activating features
The total score is then used to evaluate the structure and assign an AI limit. The Guidance’s offers worked examples to illustrate the evaluation. Additionally, the FDA list Recommended AI limits for certain hypothetical NDSRIs based on the Predicted Carcinogenicity Characterization (14) where >240 such NDSRIs are listed.
Once limits are calculated API’s should be evaluated to determine the presence of the corresponding N-Nitrosamine. Where observed levels are higher the Agency recommends manufacturers and applicants pursue mitigation efforts to reduce or remove the NDSRI(8). Where higher limits are requested the agency may request additional safety data to support alternate AI limits, this includes Mutagenicity testing. The EMA describes an enhanced Ames test conditions (1 Annex 3).
Formulation changes do not happen overnight, as these medicines cover all therapeutic areas, and for some, there are few alternatives. As a result, authorities have switched their focus to education and prevention, helping improve management and guide companies to consider better actions to ensure product safety. The document (7) provides significant details on nitrosamines in order to help the industry understand the conditions of their formation and strategies to aid prevention.
Furthermore, the FDA has encouraged companies to exploit these and other innovative solutions, supporting meeting requests to review strategies to avoid/eliminate nitrosamines, and encouraging companies to submit their formulation changes through supplements and amendments.
The requirements align with our own expertise for the testing of:
- Nitrosamines, specified or drug-related
- Nitrite testing in materials within the range of 10 50ppb
- Formulation development with an excipient compatibility
- Forced degradation studies to assess the impact of change in product stability.
We can help you assess risks and manage materials so that you can keep your products on the market and support patient safety. See Table 2 for timelines to complete evaluations and submit updates to authorities.
Method development and validation by SGS
SGS has already successfully applied its network of specialized laboratories to tackle the analytical challenges associated with a range of these somewhat unique impurities.
A coordinated regulatory strategy to address potential nitrosamine contamination in existing and new drug products has been established and includes an initial risk assessment followed by appropriate testing and mitigation of the manufacturing processes. While much attention was initially focused on traditional pharmaceutical products, in 2020 the EMA published an assessment report that directed all medicinal products, including biologics, into the scope of nitrosamine risk assessment.
Other regulatory agencies such as Health Canada, Swiss Medic, and ANVISA have now also emulated this guidance.
The overall portfolio of services provided by SGS is well-equipped to support all aspects of these regulatory strategies, from risk assessment and analytical development to evaluation and routine monitoring.

References
- EMA questions-answers for marketing authorisation holders opinion-article-53-nitrosamine-impurities-human-medicinal-products
- FDA Control of Nitrosamine Impurities in Human Drugs
- MHRA Medicines: Marketing Authorisation Holders' submission of Nitrosamine risk evaluation, risk assessment and confirmatory testing
- Health Canada Nitrosamine Impurities in Medications Guidance
- TGA - Nitrosamine impurities in medicines - Information for sponsors and manufacturers
- Japan - Self-inspection on Risks of Contamination with Nitrosamines in Drugs
- The Journal of Pharmaceutical Sciences “ The Landscape of Potential Small and Drug Substance Related Nitrosamines in Pharmaceuticals. In press and J.Pharm Sci article to be published and Open Access Published: November 16, 2022 DOI: https://doi.org/10.1016/j.xphs.2022.11.013
- FDA Updates on possible risk mitigation strategies to reduce the risk of nitrosamine drug substance-related impurities
- AAM/CHPA/PhRMA Questions for May 4th FDA-Industry Meeting to Discuss Nitrosamine Impurities in Pharmaceutical
- Control of Nitrosamine Impurities in Human Drugs, Guidance for Industry, USFDA (2021)
- Recommended Acceptable Intake Limits for Nitrosamine Drug Substances (NDSRIs) Guidance Aug 2023
- Updated Information | Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities (NDSRIs)
- Li Y, Hecht SS, 2022. Metabolic Activation and DNA Interactions of Carcinogenic N-Nitrosamines to Which Humans Are Commonly Exposed, Int J Mol Sci, 23:4559.
- Recommended AI Limits for Certain NDSRIs
16th Floor, Block A, No.73 Fucheng Road, Century Yuhui Mansion,
Beijing, Haidian District, China